Programming Assignment 1Date Posted: September 10
|
||||
| Background Information | ||||
|
In 1866 an Austrian monk named Gregor Mendel published a paper in
Proceedings of the Natural Society of Brunn, in which he described
the laws of genetics which he had formulated from his work cultivating,
cross-breeding, and testing thousands of pea plants (Pisum sativum).
Some of the traits he studied were plant stature (tall vs. dwarf), seed color
(green vs. yellow), and seed pod texture (smooth vs. wrinkled). While
getting little attention from the scientific community at first, his work later
became the foundation for the entire science of genetics and today Mendel is
known as the "father of modern genetics." Mendel showed that for any particular trait, such as plant stature, there were actually two genes forming a matched gene pair to determine the trait. Each of the genes in the pair could be either of two types. One gene type produced tall plants. This he represented with a T. The other produced dwarf plants and he represented this with a t. Thus, to represent the genotype or genetic makeup of a plant with regard to stature he could use, TT, Tt, or tt. He found that if a plant had at least one "tall" gene (represented as TT or Tt) it was always tall. This gene type he called dominant. Only when the genotype for a plant’s stature was represented as tt was it a dwarf plant. This gene type he called recessive. Mendel came to this conclusion when he found that if he crossed a plant that always produced tall offspring, i.e. was pure-bred tall, with a plant that always produced dwarf offspring, i.e. was pure-bred dwarf, he got not medium sized plants but all tall plants. And, when he crossed these plants he got both tall and dwarf plants in a ratio of three tall to one dwarf. Mendel said that organisms have genes for traits in pairs. That when reproducing each parent plant contributes one of their pair of genes for each trait to each of the offspring. Which gene of the pair is contributed is purely random. This is easy to see if we look at a diagram representing the possible combinations. On the left we see the possible combinations when crossing a pure-bred tall (also called homozygous tall) with a pure-bred dwarf (also called homozygous dwarf) plant. All the offspring will be hybrid tall (also called heterozygous tall). When two of these hybrid plants are crossed we see that the phenotypes (visible traits) give a ratio of tall to dwarf plants of 3:1. The possible genotype combinations give a ratio of one homozygous tall to two heterozygous tall to one homozygous dwarf. 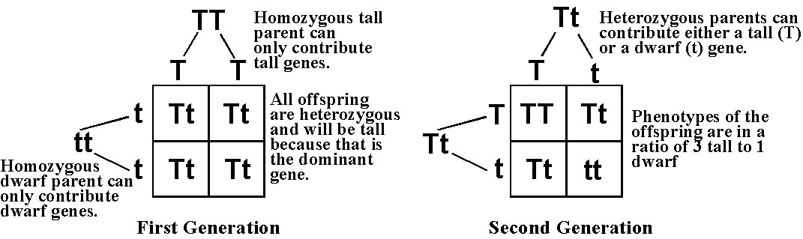 |
||||
| Your Assignment | ||||
|
You are to write a simulation program to
demonstrate the processes of inheritance incorporating the Mendelian laws
of genetics. The design and implementation of this program must follow object oriented design principles. Opportunity will be provided for you to question the customer's representative (the instructor) in class for more details. Following is a list of requirements for this simulation.
|
||||
| Deliverables | ||||
|
These products as specified below shall be delivered
electronically via e-mail to the instructor. Preliminary Class Diagram -- The class diagram shall be drawn using standard UML notation and shall show all of the classes to be implemented in the software and their relationships (dependencies, associations, generalizations, realizations, etc.) The PCD shall be submitted for instructor approval NLT (Not Later Than) Thursday, September 19. Class Outline -- The class outline shall list all proposed variables and functions in each proposed class with a brief description of what each does. The class outline shall be submitted for instructor approval NLT (Not Later Than) Thursday, September 26. Functionality Outline -- The functionality outline shall be an outline which will show the step-by-step functionality of the program. This should be taken out to a fair amount of detail. The functionality outline shall be submitted for instructor approval NLT (Not Later Than) Thursday, October 3. Final Project -- The entire software project (compatible with Microsoft Visual Studio 2012 or 2015) shall be compressed into a zip file and submitted for instructor approval NLT Tuesday, October 15. Just turning in your source files is not acceptable. |
||||
|
We will have several class periods in which we will meet as a team to discuss and plan this project. We will be doing a lot of brainstorming and planning together, but remember that each person is responsibility for implementing the final design on his/her own. |
||||
|
To download a sample executable as well as a data file and a parser for the data file click here. |
||||
|
Input File Format |
||||
This file will be provided by the instructor. It will be in a modified
XML format. A data parser class (GeneticsSimDataParser.h and .cpp)
will be provided for reading, parsing, and providing data from
the file. Below is a sample of the type of data file that
will be used by the simulation.<!-- Sample data file for use in the Mendelian Genetics Simulation Program 1 --> <!-- Note: This file is not fully compatible with XML standards, but close enough --> <!-- for the purposes of this project --> <MENDELIAN_GENETICS_SIM> <ORGANISM> <GENUS> Pisum </GENUS> <SPECIES> Sativum </SPECIES> <COMMON_NAME> Pea Plant <COMMON_NAME> </ORGANISM> <GENES> <GENE> <GENE_TRAIT> Plant Stature </GENE_TRAIT> <DOMINANT_ALLELE> Tall </DOMINANT_ALLELE> <DOMINANT_SYMBOL> T </DOMINANT_SYMBOL> <RECESSIVE_ALLELE> Dwarf </RECESSIVE_ALLELE> <RECESSIVE_SYMBOL> t </RECESSIVE_SYMBOL> </GENE> <GENE> <GENE_TRAIT> Seed Texture </GENE_TRAIT> <DOMINANT_ALLELE> Wrinkled </DOMINANT_ALLELE> <DOMINANT_SYMBOL> W </DOMINANT_SYMBOL> <RECESSIVE_ALLELE> Smooth </RECESSIVE_ALLELE> <RECESSIVE_SYMBOL> w </RECESSIVE_SYMBOL> </GENE> <GENE> <GENE_TRAIT> Seed Color </GENE_TRAIT> <DOMINANT_ALLELE> Green </DOMINANT_ALLELE> <DOMINANT_SYMBOL> S </DOMINANT_SYMBOL> <RECESSIVE_ALLELE> Yellow </RECESSIVE_ALLELE> <RECESSIVE_SYMBOL> s </RECESSIVE_SYMBOL> </GENE> <GENE> <GENE_TRAIT> Flower Color </GENE_TRAIT> <DOMINANT_ALLELE> Purple </DOMINANT_ALLELE> <DOMINANT_SYMBOL> C </DOMINANT_SYMBOL> <RECESSIVE_ALLELE> White </RECESSIVE_ALLELE> <RECESSIVE_SYMBOL> c </RECESSIVE_SYMBOL> </GENE> </GENES> <PARENTS> <PARENT> <GENOTYPE> Tt Ww Ss Cc </GENOTYPE> </PARENT> <PARENT> <GENOTYPE> Tt Ww Ss Cc </GENOTYPE> </PARENT> </PARENTS> <MENDELIAN_GENETICS_SIM> |
||||
| Output Format | ||||
The output printed on the screen shall contain the results of an experimental run in the following format:
Master Genes:
Trait Name: Plant Stature
Dominant Name: Tall(T)
Recessive Name: Dwarf(t)
Trait Name: Seed Texture
Dominant Name: Wrinkled(W)
Recessive Name: Smooth(w)
Trait Name: Seed Color
Dominant Name: Green(S)
Recessive Name: Yellow(s)
Trait Name: Flower Color
Dominant Name: Purple(C)
Recessive Name: White(c)
Sim parent 1
Organism genus-species: Pisum Sativum
Common name: Pea Plant
Genes:
Gene type = Plant Stature
Genotype = Tt
Gene type = Seed Texture
Genotype = Ww
Gene type = Seed Color
Genotype = Ss
Gene type = Flower Color
Genotype = Cc
Sim parent 2
Organism genus-species: Pisum Sativum
Common name: Pea Plant
Genes:
Gene type = Plant Stature
Genotype = Tt
Gene type = Seed Texture
Genotype = Ww
Gene type = Seed Color
Genotype = Ss
Gene type = Flower Color
Genotype = Cc
How many offspring do you want to generate? (Type the number then press Enter)
==>1000
======================= Results of this Run =======================
Gene: Plant Stature
251 homozygous dominant (Tall TT)
498 heterozygous dominant (Tall Tt)
251 homozygous recessive (Dwarf tt)
Gene: Seed Texture
240 homozygous dominant (Wrinkled WW)
498 heterozygous dominant (Wrinkled Ww)
262 homozygous recessive (Smooth ww)
Gene: Seed Color
242 homozygous dominant (Green SS)
500 heterozygous dominant (Green Ss)
258 homozygous recessive (Yellow ss)
Gene: Flower Color
221 homozygous dominant (Purple CC)
526 heterozygous dominant (Purple Cc)
253 homozygous recessive (White cc)
You can earn an extra 5 points on this programming assignment
if you also include the following information...
All occurring genotypes with the count of each.
Genotype = TT WW SS CC Offspring count = 4
Genotype = TT WW SS Cc Offspring count = 9
Genotype = TT WW Ss CC Offspring count = 8
Genotype = TT WW Ss Cc Offspring count = 12
Genotype = TT Ww SS CC Offspring count = 2
*** All other occurring genotypes are listed here but not
shown on this web page to save space. See the demonstration
of programming assignment 1.
Genotype = tt ww Ss Cc Offspring count = 16
Genotype = tt ww SS cc Offspring count = 2
Genotype = tt ww Ss cc Offspring count = 7
Genotype = tt ww ss Cc Offspring count = 8
Genotype = tt ww ss cc Offspring count = 2
|
||||
| The data parser contains a function (bool GeneticsSimDataParser::getParentGenotype(char *genotype)). Which takes a single argument of a character array. This should be at least 32 characters in length. On return it will contain a null terminated string defining the genotype of one of the parent organisms, e.g. "Tt Ww Ss Cc". A second call to this function will get the genotype of the other parent organism. Below is a simple algorithm for parsing the characters from a parent genotype. | ||||
Suppose you have a parent genotype given by the string "Tt Ww Ss Cc". This
code should parse out all the letters.
// Assume char line[32] holds a string of paired letters
char *cptr;
char ch1, ch2; // Characters of a pair
cptr = line; // Set pointer to first letter in string
while(*cptr != '\0') // while we have not reached the null terminator
{
ch1 = *cptr; // Read first character of a pair
cptr++; // Increment to next letter
ch2 = *cptr; // Read second character of a pair
cptr++; // Increment to next space or null terminator
if(*cptr == ' ') // If it's a space...
cptr++; // ...increment to next character
// Note: in the above if statement, if *cptr is not a space
// then it must be the null terminator at the end of the
// string so we don't want to increment past it or the while
// loop will not terminate properly.
// Create a new gene for this pair of characters
Gene *g = new Gene();
if(ch1 < ch2) // If ch1 is a capital letter and ch2 is lower case
{ // Note: Capital letters have a lower ASCII value than lower case
g->setAllele1(ch1); // If the gene pair is "Tt" we want 'T' in
g->setAllele2(ch2); // the first allele and 't' in the second.
}
else // Either both are the same or ch2 is the capital
{
g->setAllele1(ch2);
g->setAllele2(ch1);
}
// Call the appropriate function here to add the new gene
// to the current parent organism
}
|
||||